




Ouro-Tools - long-read scRNA-seq toolkit
Ouro-Tools is a novel, comprehensive computational pipeline for long-read scRNA-seq with the following key features. Ouro-Tools (1) normalizes mRNA size distributions and (2) detects mRNA 7-methylguanosine caps to integrate multiple single-cell long-read RNA-sequencing experiments across modalities and characterize full-length transcripts, respectively.

-
step 4) Biological full-length molecule identification module
-
wrap-up) Running the entire pipeline using a wrapper function
-
Pre-built indices of unwanted genomic sequences for pre-processing
The Ouro-Tools pipeline comprises five main modules, allowing seamless integration with existing bulk and single-cell long-read RNA-seq pipelines and tools. Every main module of Ouro-Tools utilizes efficient parallelization for compute-intensive tasks to facilitate the processing of large datasets. Additionally, each Ouro-Tools module employs filesystem-based locks for parallel processing of a large number of samples across multiple machines for scalability.
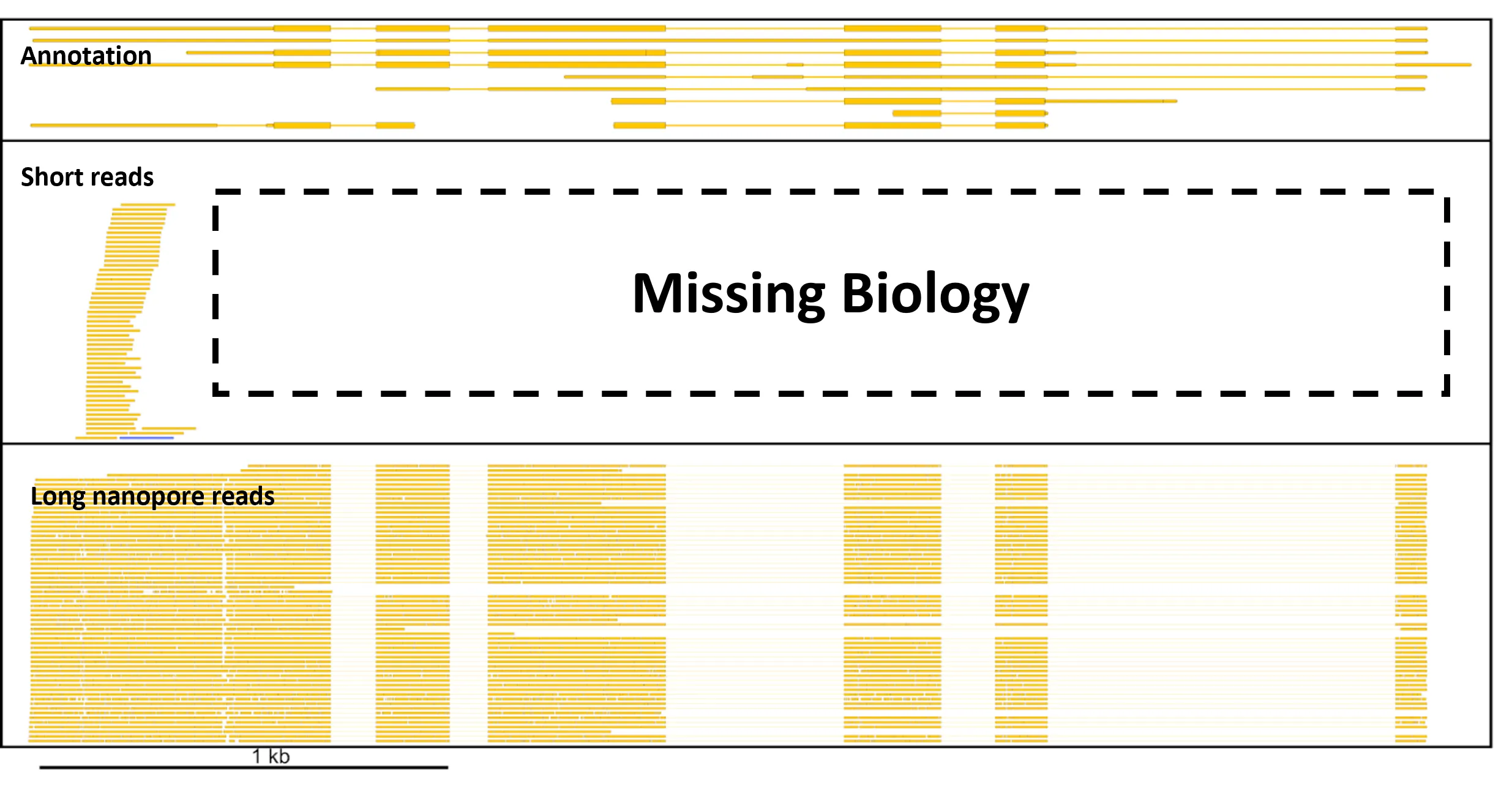
(Figure adapted from Volden & Vollmers, Genome Biol. 23:47 (2022), and made available under Creative Commons license 4.0 by Oxford Nanopore Technologies plc.)
In 2013, 2019, and 2022, “single-cell sequencing,” “single-cell multimodal omics,” and “long-read sequencing” were chosen as “Method of the Year” by Nature Methods journal, respectively, highlighting the urgent need to understand biology at the resolution of individual cells and individual biological molecules. Long-read scRNA-seq is a method that combines the single-cell RNA sequencing and long-read sequencing (Nanopore and PacBio) methods.
The latest stable version of Ouro-Tools is available in PyPI, BioConda, and BioContainers.
PyPI Installation (as a Python package)
pip install ourotoolsBioconda Installation (as an Anaconda package)
conda install bioconda::ourotoolsBioContainers Installation (as a Docker container)
# Download the latest Ouro-Tools Docker image from BioContainers
docker pull quay.io/biocontainers/ourotools:0.2.8--pyhdfd78af_0
# Run the Ouro-Tools Docker image
# please change '/your/local/folder' to the local path containing the input files
docker run -v /your/local/folder:/data -it quay.io/biocontainers/ourotools:0.2.8--pyhdfd78af_0Install the latest (but possibly unstable) version via GitHub
git clone https://github.com/ahs2202/ouro-tools.git
cd ouro-tools
pip install .Test the installation
Ouro-Tools can be used in command line, in a Python script, or in an interactive Python interpreter (e.g., Jupyter Notebook).
To print the command line usage example of each module from the bash shell, please type the following command.
Bash shell
ourotools LongFilterNSplit -hIPython environment (Jupyter Notebook)
ourotools.LongFilterNSplit?Each toy dataset contains a subsampled long-read sequencing (ONT R10.4.1) result of an Ouro-Seq library (please check our BioRxiv pre-print for more details). 3 cell types (100 cells are sampled for each cell type) and 3 chromosomes are selected for subsampling. Additionally, the artifact molecules were subsampled and included in the toy dataset.
# download toy datasets from mouse ovary and testis
wget https://ouro-tools.s3.amazonaws.com/tutorial/mOvary.subsampled.fastq.gz
wget https://ouro-tools.s3.amazonaws.com/tutorial/mTestis2.subsampled.fastq.gz Alternatively, you can download directly using your browser using the following links: mOvary and mTestis
import ourotools
# global multiprocessing settings
ourotools.bk.int_max_num_batches_in_a_queue_for_each_worker = 1 # [NOTE] For WSL, changing this variable to 1 is necessary to prevent deadlock(s) during IPC.
n_workers = 2 # employ 2 workers (since there are two samples, 2 workers are sufficient)
n_threads_for_each_worker = 8 # use 8 CPU cores for each worker
# datasets-specific setting
path_folder_data = '/home/project/Single_Cell_Full_Length_Atlas/data/pipeline/20220331_Ouroboros_Project/pipeline/20230208_Mouse_Long_Read_Single_Cell_Atlas/pipeline/20230811_mouse_long_read_single_cell_atlas_v202308/tutorial_data/20240728_ovary_testis_tutorial/'
l_name_sample = [
'mOvary.subsampled',
'mTestis2.subsampled',
]
# scRNA-seq technology-specific settings
path_file_valid_barcode_list = '/home/project/Single_Cell_Full_Length_Atlas/data/pipeline/20210728_development_ouroboros_qc/example/3M-february-2018.txt.gz' # GEX v3 CB
# species-specific settings
path_file_minimap_index_genome = '/home/shared/ensembl/Mus_musculus/index/minimap2/Mus_musculus.GRCm38.dna.primary_assembly.k_14.idx'
path_file_minimap_splice_junction = '/home/shared/ensembl/Mus_musculus/Mus_musculus.GRCm38.102.paftools.bed'
path_file_minimap_unwanted = '/home/project/Single_Cell_Full_Length_Atlas/data/accessory_data/cDNA_depletion/index/minimap2/MT_and_rRNA_GRCm38.fa.ont.mmi'
path_folder_count_module_index = '/home/project/Single_Cell_Full_Length_Atlas/data/pipeline/20211116_ouroboros_short_read_public_data_mining/scarab_annotations/Mus_musculus.GRCm38.102.v0.2.4/' # path to the Ouro-Tools count module indexTo find the barcode whitelist specific to your scRNA-seq experiment, please refer to the official 10x Genomics article. Pre-built Ouro-Tools count module index can be downloaded here. Pre-built indices of unwanted sequences (ribosomal DNA repeats and mitochondrial DNAs) can be downloaded here.
# run LongFilterNSplit
ourotools.LongFilterNSplit(
path_file_minimap_index_genome = path_file_minimap_index_genome,
l_path_file_minimap_index_unwanted = [ path_file_minimap_unwanted ],
l_path_file_fastq_input = list( f"{path_folder_data}{name_sample}.fastq.gz" for name_sample in l_name_sample ),
l_path_folder_output = list( f"{path_folder_data}LongFilterNSplit_out/{name_sample}/" for name_sample in l_name_sample ),
int_num_samples_analyzed_concurrently = n_workers,
n_threads = n_workers * n_threads_for_each_worker,
)As the first module of the Ouro-Tools pipeline, the raw long-read pre-processing module LongFilterNSplit has a dual function for (1) providing comprehensive quality control metrics of a long-read scRNA-seq experiment and (2) pre-processing of raw long-read sequencing data for the downstream analysis.

According to the classification results, cDNA molecules are organized into separate output FASTQ files. For the cDNA molecules that contains a single (external or internal) poly(A) tail, the read is re-oriented so that it has the same orientation as its original mRNA transcript, with the poly(A) tail at its 3’ end; the resulting long-reads of cDNAs can be utilized for strand-specific long-read RNA-seq analysis.
# align using minimap2 (require that minimap2 executable can be found in PATH)
# below is a wrapper function for minimap2
ourotools.Workers(
ourotools.ONT.Minimap2_Align, # function to deploy
int_num_workers_for_Workers = n_workers, # create 'n_workers' number of workers
# below are arguments for the function 'ourotools.ONT.Minimap2_Align'
path_file_fastq = list( f"{path_folder_data}LongFilterNSplit_out/{name_sample}/aligned_to_genome__non_chimeric__poly_A__plus_strand.fastq.gz" for name_sample in l_name_sample ),
path_folder_minimap2_output = list( f"{path_folder_data}minimap2_bam_genome/{name_sample}/" for name_sample in l_name_sample ),
path_file_junc_bed = path_file_minimap_splice_junction,
path_file_minimap2_index = path_file_minimap_index_genome,
n_threads = n_threads_for_each_worker,
)Minimap2 can be used for annotation-guided alignment based on the transcript annotations prepared by the researcher. Here, the reference annotation from Ensembl (Ensembl release 102) was utilized.
# run LongExtractBarcodeFromBAM
l_path_folder_barcodedbam = list( f"{path_folder_data}LongExtractBarcodeFromBAM_out/{name_sample}/" for name_sample in l_name_sample )
ourotools.LongExtractBarcodeFromBAM(
path_file_valid_cb = path_file_valid_barcode_list,
l_path_file_bam_input = list( f"{path_folder_data}minimap2_bam_genome/{name_sample}/aligned_to_genome__non_chimeric__poly_A__plus_strand.fastq.gz.minimap2_aligned.bam" for name_sample in l_name_sample ),
l_path_folder_output = l_path_folder_barcodedbam,
int_num_samples_analyzed_concurrently = n_workers,
n_threads = n_workers * n_threads_for_each_worker,
)The barcode extraction module LongExtractBarcodeFromBAM identifies cell barcode (CB) and unique molecular identifier (UMI) sequences for each read and exports the results as a “barcoded” BAM file, a BAM file containing corrected CB and UMI sequences for each read using the predefined SAM tags.

# run full-length ID module
# survey 5' sites for each sample
ourotools.LongSurvey5pSiteFromBAM(
l_path_folder_input = l_path_folder_barcodedbam,
int_num_samples_analyzed_concurrently = n_workers,
n_threads = n_workers * n_threads_for_each_worker,
)
# combine 5' site profiles across samples and classify each 5' profile
ourotools.LongClassify5pSiteProfiles(
l_path_folder_input = l_path_folder_barcodedbam,
path_folder_output = f"{path_folder_data}LongClassify5pSiteProfiles_out/",
n_threads = n_threads_for_each_worker,
)
# append 5' site classification results to each BAM file
ourotools.LongAdd5pSiteClassificationResultToBAM(
path_folder_input_5p_sites = f'{path_folder_data}LongClassify5pSiteProfiles_out/',
l_path_folder_input_barcodedbam = l_path_folder_barcodedbam,
int_num_samples_analyzed_concurrently = n_workers,
n_threads = n_workers * n_threads_for_each_worker,
)
# filter artifact reads from each BAM file
ourotools.Workers(
ourotools.FilterArtifactReadFromBAM, # function to deploy
int_num_workers_for_Workers = n_workers, # create 'n_workers' number of workers
# below are arguments for the function 'ourotools.FilterArtifactReadFromBAM'
path_file_bam_input = list( f'{path_folder_data}LongExtractBarcodeFromBAM_out/{name_sample}/5pSiteTagAdded/barcoded.bam' for name_sample in l_name_sample ),
path_folder_output = list( f'{path_folder_data}LongExtractBarcodeFromBAM_out/{name_sample}/5pSiteTagAdded/FilterArtifactReadFromBAM_out/' for name_sample in l_name_sample ),
)The biological full-length identification module collects the lengths of guanosine homopolymers at the 5’ ends of cDNAs to identify genuine TSSs that produce capped mRNAs, depleting truncated cDNA molecules in silico. The module is implemented as a workflow consisting of LongSurvey5pSiteFromBAM, LongClassify5pSiteProfiles, LongAdd5pSiteClassificationResultToBAM, and FilterArtifactReadFromBAM.

# run mRNA size distribution normalization module
# survey the size distribution of full-length mRNAs for each sample
l_full_length_bam = list( f'{path_folder_data}LongExtractBarcodeFromBAM_out/{name_sample}/5pSiteTagAdded/FilterArtifactReadFromBAM_out/valid_3p_valid_5p.bam' for name_sample in l_name_sample )
ourotools.Workers(
ourotools.LongSummarizeSizeDistributions,
int_num_workers_for_Workers = n_workers, # create 'n_workers' number of workers
path_file_bam_input = l_full_length_bam,
path_folder_output = list( f'{path_folder_data}LongExtractBarcodeFromBAM_out/{name_sample}/5pSiteTagAdded/FilterArtifactReadFromBAM_out/valid_3p_valid_5p.LongSummarizeSizeDistributions_out/' for name_sample in l_name_sample ),
)
# normalize size distributions
path_folder_size_norm = f"{path_folder_data}LongCreateReferenceSizeDistribution_out/"
ourotools.LongCreateReferenceSizeDistribution(
l_path_file_distributions = list( f'{path_folder_data}LongExtractBarcodeFromBAM_out/{name_sample}/5pSiteTagAdded/FilterArtifactReadFromBAM_out/valid_3p_valid_5p.LongSummarizeSizeDistributions_out/dict_arr_dist.pkl' for name_sample in l_name_sample ),
l_name_file_distributions = l_name_sample,
path_folder_output = path_folder_size_norm,
float_max_ratio_to_arr_dist_guassian_filter_min_sigma_for_dynamic_gaussian_filter_selection = 2,
float_sigma_gaussian_filter_min = 8,
int_min_total_read_count_for_a_peak = 30 ,
)
# based on the output, set the confident size range
str_confident_size_range = ourotools.get_confident_size_range( path_folder_size_norm )The size distribution normalization module is implemented using the LongSummarizeSizeDistributions and LongCreateReferenceSizeDistribution workflows. First, using the LongSummarizeSizeDistributionsworkflow, a full-length, UMI-deduplicated cDNA size distribution is obtained from the valid_3p_valid_5p barcoded BAM file (representing in vivo full-length mRNAs) for each sample. Next, the reference mRNA size distribution is constructed for all the samples using the LongCreateReferenceSizeDistribution workflow.

# run the single-cell count module
ourotools.LongExportNormalizedCountMatrix(
path_folder_ref = path_folder_count_module_index,
l_path_file_bam_input = l_full_length_bam,
l_path_folder_output = list( f'{path_folder_data}LongExportNormalizedCountMatrix_out/{name_sample}/' for name_sample in l_name_sample ),
l_name_distribution = l_name_sample,
path_folder_reference_distribution = path_folder_size_norm,
l_str_l_t_distribution_range_of_interest = [ ','.join( [ "raw", str_confident_size_range ] ) ],
flag_enforce_transcript_start_site_matching_for_long_read_during_realignment = True,
flag_enforce_transcript_end_site_matching_for_long_read_during_realignment = True,
)The single-cell long-read count module LongExportNormalizedCountMatrix is largely composed of three parts: constructing an index (only required once for each set of genes, transcripts, repeat elements, regulatory elements and the reference genome), assigning each read to various buckets (each bucket represent one of the genes, transcripts, exons, splice junctions, TEs, tCREs, and individual genomic tiles), and exporting a size distribution-normalized count matrix for each bucket (later these count matrixes are combined into a single size distribution-normalized count matrix as an output).
# TBD# version 2025-01-25 by Hyunsu An @ GIST-FGL
import ourotools
ourotools.bk.int_max_num_batches_in_a_queue_for_each_worker = 1 # [NOTE] For WSL, changing this variable to 1 is necessary to prevent deadlock(s) during IPC.
ourotools.run_pipeline(
# dataset setting
path_folder_data = '/home/project/Single_Cell_Full_Length_Atlas/data/pipeline/20220331_Ouroboros_Project/pipeline/20230208_Mouse_Long_Read_Single_Cell_Atlas/pipeline/20230811_mouse_long_read_single_cell_atlas_v202308/tutorial_data/20240813_ovary_testis_tutorial2/',
l_name_sample = [
'mOvary.subsampled',
'mTestis2.subsampled',
],
# scRNA-seq technology-specific
path_file_valid_barcode_list = '/home/project/Single_Cell_Full_Length_Atlas/data/pipeline/20210728_development_ouroboros_qc/example/3M-february-2018.txt.gz', # GEX v3 CB
# species-specific settings
path_file_minimap_index_genome = '/home/shared/ensembl/Mus_musculus/index/minimap2/Mus_musculus.GRCm38.dna.primary_assembly.k_14.idx',
path_file_minimap_splice_junction = '/home/shared/ensembl/Mus_musculus/Mus_musculus.GRCm38.102.paftools.bed',
path_file_minimap_unwanted = '/home/project/Single_Cell_Full_Length_Atlas/data/accessory_data/cDNA_depletion/index/minimap2/MT_and_rRNA_GRCm38.fa.ont.mmi',
path_folder_count_module_index = '/home/project/Single_Cell_Full_Length_Atlas/data/pipeline/20211116_ouroboros_short_read_public_data_mining/scarab_annotations/Mus_musculus.GRCm38.102.v0.2.4/', # path to the Ouro-Tools reference
# run setting
n_workers = 2, # employ 2 workers (since there are two samples, 2 workers are sufficient)
n_threads_for_each_worker = 8, # use 8 CPU cores for each worker
# additional settings
args = dict(
LongCreateReferenceSizeDistribution = dict(
float_max_ratio_to_arr_dist_guassian_filter_min_sigma_for_dynamic_gaussian_filter_selection = 2,
float_sigma_gaussian_filter_min = 8,
int_min_total_read_count_for_a_peak = 30 ,
),
LongExportNormalizedCountMatrix = dict(
flag_enforce_transcript_start_site_matching_for_long_read_during_realignment = True,
flag_enforce_transcript_end_site_matching_for_long_read_during_realignment = True,
),
),
)The pre-built indices of unwanted sequences can be downloaded using the following links:
Human (GRCh38) : Minimap2-index-file, FASTA-file, GTF-file
Mouse (GRCm38) : Minimap2-index-file, FASTA-file, GTF-file
The single-cell count module of Ouro-Tools utilizes genome, transcriptome, and gene annotations to assign reads to genes, isoforms, and genomic bins (tiles across the genome). The index building process is automatic; there is no needs to run a separate command in order to build the index. Once Ouro-Tools processes these information before analyzing an input BAM file(s), the program saves an index in order to load the information much faster next time.
We recommends using Ensembl reference genome, transcriptome, and gene annotations of the same version (release number).
pre-built index can be downloaded using the following links (should be extracted to a folder using tar -xf command):
Human (GRCh38, Ensembl version 105)
Mouse (GRCm39, Ensembl version 105)
Mouse (GRCm38, Ensembl version 102)
Zebrafish (GRCz11, Ensembl version 104)
Thale cress (TAIR10, Ensembl Plant version 56)
An Ouro-Tools index can be built on-the-fly from the input genome, transcriptome, and gene annotation files. For example, below are the list of files that were used for the pre-built Ouro-Tools index "Human (GRCh38, Ensembl version 105)".
required annotations (Ensemble version 105):
- path_file_fa_genome : https://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
- A genome FASTA file. Either gzipped or plain FASTA file can be accepted.
- path_file_gtf_genome : https://ftp.ensembl.org/pub/release-105/gtf/homo_sapiens/Homo_sapiens.GRCh38.105.gtf.gz
- A GTF file. Either gzipped or plain GTF file can be accepted. Currently GFF3 format files are not supported.
- Following arguments can be used to set attribute names for identifying gene and transcript annotations in its attributes column.
- str_name_gtf_attr_for_id_gene : (default: 'gene_id')
- str_name_gtf_attr_for_name_gene : (default: 'gene_name')
- str_name_gtf_attr_for_id_transcript : (default: 'transcript_id')
- str_name_gtf_attr_for_name_transcript : (default: 'transcript_name')
- An example of GTF annotation file for gene annotations:
1 ensembl_havana gene 1211340 1214153 . - . gene_id "ENSG00000186827"; gene_version "11"; gene_name "TNFRSF4"; gene_source "ensembl_havana"; gene_biotype "protein_coding";
1 ensembl_havana transcript 1211340 1214153 . - . gene_id "ENSG00000186827"; gene_version "11"; transcript_id "ENST00000379236"; transcript_version "4"; gene_name "TNFRSF4"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; transcript_name "TNFRSF4-201"; transcript_source "ensembl_havana"; transcript_biotype "protein_coding"; tag "CCDS"; ccds_id "CCDS11"; tag "basic"; transcript_support_level "1 (assigned to previous version 3)";
1 ensembl_havana exon 1213983 1214153 . - . gene_id "ENSG00000186827"; gene_version "11"; transcript_id "ENST00000379236"; transcript_version "4"; exon_number "1"; gene_name "TNFRSF4"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; transcript_name "TNFRSF4-201"; transcript_source "ensembl_havana"; transcript_biotype "protein_coding"; tag "CCDS"; ccds_id "CCDS11"; exon_id "ENSE00001832731"; exon_version "2"; tag "basic"; transcript_support_level "1 (assigned to previous version 3)";
- path_file_fa_transcriptome : https://ftp.ensembl.org/pub/release-105/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz
- A transcriptome FASTA file. Either gzipped or plain FASTA file can be accepted.
-
path_file_tsv_repeatmasker_ucsc : Table Browser (ucsc.edu) [click "get output" to download the annotation]
- repeat masker annotations from the UCSC Table Browser
-
path_file_gff_regulatory_element : https://ftp.ensembl.org/pub/current_regulation/homo_sapiens/homo_sapiens.GRCh38.Regulatory_Build.regulatory_features.20221007.gff.gz
-
The latest regulatory build from Ensembl.
-
Annotations from other sources, or custom annotations can be used. Currently only the GFF3 file format is supported (with .gff extension).
-
The following argument can be used to set the attribute name for identifying regulatory region
- str_name_gff_attr_id_regulatory_element : (default: 'ID')
-
An example of GFF annotation file for regulatory elements:
-
18 Regulatory_Build enhancer 35116801 35120999 . . . ID=enhancer:ENSR00000572865;bound_end=35120999;bound_start=35116801;description=Predicted enhancer region;feature_type=Enhancer
8 Regulatory_Build TF_binding_site 37967115 37967453 . . . ID=TF_binding_site:ENSR00001137252;bound_end=37967531;bound_start=37966339;description=Transcription factor binding site;feature_typ
6 Regulatory_Build enhancer 90249202 90257999 . . . ID=enhancer:ENSR00000798348;bound_end=90257999;bound_start=90249202;description=Predicted enhancer region;feature_type=Enhancer
3 Regulatory_Build CTCF_binding_site 57689401 57689600 . . . ID=CTCF_binding_site:ENSR00000687477;bound_end=57689600;bound_start=57689401;description=CTCF binding site;feature_type=CTCF
| SAM tag name | data type | Description | Module name |
|---|---|---|---|
| CB | Z | the corrected cell barcode sequence | Barcode Extraction |
| UB | Z | the corrected UMI sequence after the UMI clustering process | Barcode Extraction |
| UR | Z | the uncorrected UMI sequence before the UMI clustering process | Barcode Extraction |
| XR | i | the number of errors for identification of R1 adapter (marks the 3’ end of cDNA). -1 indicates that the adapter was not identified | Barcode Extraction |
| XT | i | the number of errors for identification of TSO adapter (marks the 5’ end of cDNA). -1 indicates that the adapter was not identified | Barcode Extraction |
| CU | Z | the uncorrected raw CB-UMI sequence | Barcode Extraction |
| IA | i | the length of detected internal poly(A) tract on the genome | Barcode Extraction |
| LE | i | the total number of genome-aligned base pairs | Barcode Extraction |
| AG | i | the number of consecutive G nucleotides, starting from the 5’ site in the aligned region of the read | Full-Length Identification |
| UG | i | the number of consecutive G nucleotides, starting from the 5’ site in the unaligned region of the read (soft-clipped sequence) | Full-Length Identification |
| VS | i | “1” if the 5’ site is identified as a valid transcript start site (TSS), “0” if the 5’ site is identified as an invalid TSS, representing 5’ sites of the PCR/RT artifacts (including 5p degradation products of full-length transcripts) | Full-Length Identification |
| AU | i | the inferred number of unreferenced G nucleotides aligned to the genome | Full-Length Identification |
| XC | i | the bitwise flags (see the table below for more details) | Single-Cell Count |
| XR | Z | the repeat element ID | Single-Cell Count |
| YR | i | the total number of base pairs overlapping with the repeat element to which the read is confidently assigned | Single-Cell Count |
| XG | Z | the gene ID | Single-Cell Count |
| YG | i | the total number of base pairs overlapping with the exons of the gene to which the read is confidently assigned | Single-Cell Count |
| XP | Z | the promoter ID | Single-Cell Count |
| YX | i | the total number of base pairs overlapping with any exons that overlap with the read | Single-Cell Count |
| YF | i | the total number of base pairs overlapping with any repeat elements that overlap with the read (considering only filtered repeat elements) | Single-Cell Count |
| XE | Z | the regulatory element ID | Single-Cell Count |
| YU | i | the total number of base pairs overlapping with any repeat elements that overlap with the read (considering all repeat elements) | Single-Cell Count |
| YE | i | the total number of base pairs overlapping with any regulatory elements that overlap with the read | Single-Cell Count |
| XT | Z | the transcript ID that is uniquely assigned to the read using the re-alignment process | Single-Cell Count |
| ZF | i | the flag that indicates the read represents a full-length cDNA with valid 3’ and 5’ ends | Single-Cell Count |
| Binary flag | Feature type | Description |
|---|---|---|
| 0x1 | gene | overlaps with gene(s) |
| 0x2 | gene | gene assignment is ambiguous |
| 0x4 | gene | completely intronic reads (GEX mode specific) |
| 0x8 | gene | exonic reads (GEX mode specific) |
| 0x10 | promoter | overlaps with promoter region(s) (ATAC mode specific) |
| 0x20 | promoter | promoter assignment is ambiguous (ATAC mode specific) |
| 0x40 | repeats | overlaps with repeat element(s) |
| 0x80 | repeats | ambiguous assignment to two or more number of repeat elements |
| 0x100 | repeats | the entire length of a read overlaps with a single repeat element |
| 0x200 | regulatory | overlaps with regulatory element(s) |
| 0x400 | regulatory | overlaps with both repeat element(s) and regulatory element(s) |
| 0x800 | regulatory | overlaps exclusively with regulatory element(s) (no overlaps with repeat element) |
| 0x1000 | regulatory | ambiguous assignment to two or more number of regulatory elements |
| 0x2000 | regulatory | the entire length of a read overlaps with a single regulatory element |
Ouro-Tools was developed by Hyunsu An and Chaemin Lim at Gwangju Institute of Science and Technology under the supervision of Professor Jihwan Park.
© 2025 Functional Genomics Lab, Gwangju Institute of Science and Technology